Notice:
This lecture was delivered on May 10, 2021. Per our 3-year review policy, we’ve decided to maintain this lecture on CHOP OPEN. This content may contain information that has since been updated.
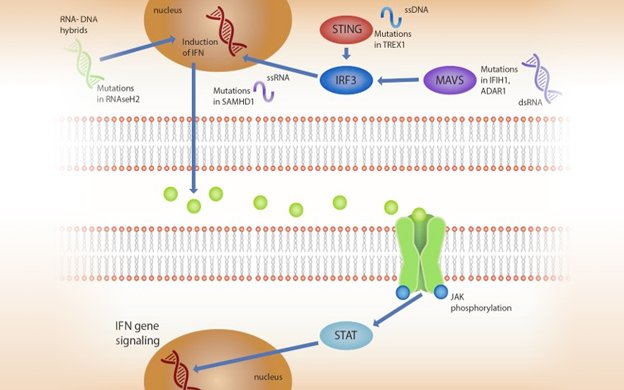
Aicardi Goutières Syndrome (AGS) is a leukodystrophy characterized by early neurologic disability. Although AGS is genetically heterogeneous, all genotypes activate a common pathway: interferon (IFN) production leading to Janus kinase (JAK) activation and transcription of IFN-stimulated genes. The neurologic impairment associated with AGS ranges from mild, stable spastic paraparesis (a mimic of cerebral palsy) to severe early onset disease (a mimic of congenital TORCH infections). Often there are subtle cues to aid in the recognition of AGS. We will discuss clinical monitoring guidelines for the AGS population to address the potential for systemic complications. While there is no approved treatment, there is great interest in targeting common steps along this autoinflammatory pathway. We will address the evidence surrounding therapeutic interventions from steroids and IVIG to reappropriation of targeted therapies such as reverse transcriptase inhibitors and janus kinase inhibition. We will discuss the common laboratory abnormalities observed during the clinical trials of baricitinib. Ultimately, this complicated disorder of autoinflammation necessitates a comprehensive plan for clinical monitoring with additional monitoring while on immunomodulatory treatments.
Learning Objectives:
- Review the early signs and symptoms of AGS to help with earlier diagnosis
- Review a strategy for comprehensive clinical management and monitoring in AGS
- Review the evidence behind emerging treatment strategies such as janus kinase inhibition
Speakers:
This seminar focuses on neurology and was delivered at a virtual event titled, “Aicardi Goutieres Syndrome: Diagnosis and Management,” on May 10, 2021.
Additional CHOP Resources:
Content Disclaimer
The Terms of Use and Privacy Policy set forth on the website of The Children’s Hospital of Philadelphia apply to any and all uses of and access to this site and the content found here.
The work presented in the presentations, videos, and other content on this site (“Presentations”) includes publicly available medical evidence, a consensus of medical practitioners, and/or opinions of individual practitioners that may differ from consensus opinions. These Presentations are intended only to provide general information and need to be adapted for each specific patient based on the practitioner’s professional judgment, consideration of any unique circumstances, the needs of each patient and their family, the availability of various resources at the health care institution where the patient is located, and other factors. The Presentations are not intended to constitute medical advice or treatment, nor should they be relied upon as such. The Presentations are not intended to create a doctor-patient relationship between/among The Children’s Hospital of Philadelphia, its physicians and the individual patients in question. The information contained in these Presentations are general in nature, and do not and are not intended to refer to specific patients.
CHOP, The Children’s Hospital of Philadelphia Foundation and its or their affiliates, the authors, presenters, practitioners, editors, and others associated with the creation of the Presentations (“CHOP”) are not responsible for errors or omissions in the Presentations; for any outcomes a patient might experience where a clinician reviewed one or more such Presentations in connection with providing care for that patient; and/or for any and all third party content on the site or in the Presentations. CHOP makes no warranty, expressed or implied, with respect to the currency, completeness, applicability or accuracy of the Presentations. Application of the information in or to a particular situation remains the professional responsibility of the practitioner who is directly treating the patient.
To the extent that the Presentations include information regarding drug dosing, in view of ongoing research, changes in government regulations and the constant flow of information relating to drug therapy and drug reactions, the viewer should not rely on the Presentation content, but rather is urged to check the package insert for each drug for indications, dosage, warnings and precautions.
Some drugs and medical devices presented in the Presentations have United States Food and Drug Administration (FDA) clearance for limited use in restricted research settings. It is the responsibility of the practitioner to ascertain the FDA status of each drug or device planned for use in their clinical practice.
You shall indemnify, defend and hold harmless CHOP, The Children’s Hospital of Philadelphia Foundation, and its/their current and former employees, officers, and agents, trustees, and their respective successors, heirs and assigns (“Indemnitees”) against any claims, liability, damage, loss or expenses (including attorneys’ fees and expenses of litigation) in connection with any claims, suits, actions, demands or judgments arising directly or indirectly out of your reference to or use of the Presentations.
The Presentations are protected by copyright laws and in some cases patent laws, and all rights are reserved under such laws. No part of the Presentations may be reproduced in any form by any means, or utilized in any other way, absent prior written permission from the copyright owner.
By starting this module, you agree to our Content Disclaimer and Terms of Service.
Course Includes
- 2 Lessons
- Course Certificate